Note
Go to the end to download the full example code. or to run this example in your browser via Binder
Producing single subject maps of seed-to-voxel correlation¶
This example shows how to produce seed-to-voxel correlation maps for a single subject based on movie-watching fMRI scans. These maps depict the temporal correlation of a seed region with the rest of the brain.
This example is an advanced one that requires manipulating the data with numpy. Note the difference between images, that lie in brain space, and the numpy array, corresponding to the data inside the mask.
Getting the data¶
We will work with the first subject of the brain development fMRI data set. dataset.func is a list of filenames. We select the 1st (0-based) subject by indexing with [0]).
from nilearn import datasets, plotting
dataset = datasets.fetch_development_fmri(n_subjects=1)
func_filename = dataset.func[0]
confound_filename = dataset.confounds[0]
[fetch_development_fmri] Dataset directory found:
/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri
[fetch_development_fmri] Dataset directory found:
/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri
[fetch_development_fmri] Dataset directory found:
/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri
Note that func_filename and confound_filename are strings pointing to files on your hard drive.
print(func_filename)
print(confound_filename)
/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_space-MNI152NLin2009cAsym_desc-preproc_bold.nii.gz
/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_desc-reducedConfounds_regressors.tsv
Time series extraction¶
We are going to extract signals from the functional time series in two steps. First we will extract the mean signal within the seed region of interest. Second, we will extract the brain-wide voxel-wise time series.
We will be working with one seed sphere in the Posterior Cingulate Cortex (PCC), considered part of the Default Mode Network.
pcc_coords = [(0, -52, 18)]
We use NiftiSpheresMasker to extract the
time series from the functional imaging within the sphere. The
sphere is centered at pcc_coords and will have the radius we pass the
NiftiSpheresMasker function (here 8 mm).
The extraction will also detrend, standardize, and bandpass filter the data. This will create a NiftiSpheresMasker object.
from nilearn.maskers import NiftiSpheresMasker
seed_masker = NiftiSpheresMasker(
pcc_coords,
radius=8,
detrend=True,
standardize_confounds=True,
low_pass=0.1,
high_pass=0.01,
t_r=dataset.t_r,
memory="nilearn_cache",
memory_level=1,
verbose=1,
)
Then we extract the mean time series within the seed region while regressing out the confounds that can be found in the dataset’s csv file
seed_time_series = seed_masker.fit_transform(
func_filename, confounds=[confound_filename]
)
\[NiftiSpheresMasker.wrapped] Finished fit
/home/runner/work/nilearn/nilearn/examples/03_connectivity/plot_seed_to_voxel_correlation.py:81: FutureWarning:
boolean values for 'standardize' will be deprecated in nilearn 0.15.0.
Use 'zscore_sample' instead of 'True' or use 'None' instead of 'False'.
________________________________________________________________________________
[Memory] Calling nilearn.maskers.base_masker.filter_and_extract...
filter_and_extract('/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_space-MNI152NLin2009cAsym_desc-preproc_bold.nii.gz',
<nilearn.maskers.nifti_spheres_masker._ExtractionFunctor object at 0x7f8101e26a40>,
{ 'allow_overlap': False,
'clean_args': None,
'clean_kwargs': {},
'detrend': True,
'dtype': None,
'high_pass': 0.01,
'high_variance_confounds': False,
'low_pass': 0.1,
'mask_img': None,
'radius': 8,
'reports': True,
'seeds': [(0, -52, 18)],
'smoothing_fwhm': None,
'standardize': False,
'standardize_confounds': True,
't_r': 2}, confounds=[ '/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_desc-reducedConfounds_regressors.tsv'], sample_mask=None, dtype=None, sklearn_output_config=None, memory=Memory(location=nilearn_cache/joblib), memory_level=1, verbose=1)
\[NiftiSpheresMasker.wrapped] Loading data from '/home/runner/work/nilearn/nilea
rn/nilearn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_space-
MNI152NLin2009cAsym_desc-preproc_bold.nii.gz'
\[NiftiSpheresMasker.wrapped] Extracting region signals
\[NiftiSpheresMasker.wrapped] Cleaning extracted signals
/home/runner/work/nilearn/nilearn/examples/03_connectivity/plot_seed_to_voxel_correlation.py:81: FutureWarning:
boolean values for 'standardize' will be deprecated in nilearn 0.15.0.
Use 'zscore_sample' instead of 'True' or use 'None' instead of 'False'.
_______________________________________________filter_and_extract - 1.6s, 0.0min
Next, we can proceed similarly for the brain-wide voxel-wise time
series, using NiftiMasker with the same input
arguments as in the seed_masker in addition to smoothing with a 6 mm kernel
from nilearn.maskers import NiftiMasker
brain_masker = NiftiMasker(
smoothing_fwhm=6,
detrend=True,
standardize_confounds=True,
low_pass=0.1,
high_pass=0.01,
t_r=dataset.t_r,
memory="nilearn_cache",
memory_level=1,
verbose=1,
)
Then we extract the brain-wide voxel-wise time series while regressing out the confounds as before
brain_time_series = brain_masker.fit_transform(
func_filename, confounds=[confound_filename]
)
\[NiftiMasker.wrapped] Loading data from '/home/runner/work/nilearn/nilearn/nile
arn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_space-MNI152N
Lin2009cAsym_desc-preproc_bold.nii.gz'
\[NiftiMasker.wrapped] Computing mask
________________________________________________________________________________
[Memory] Calling nilearn.masking.compute_background_mask...
compute_background_mask('/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_space-MNI152NLin2009cAsym_desc-preproc_bold.nii.gz', verbose=0)
__________________________________________compute_background_mask - 0.2s, 0.0min
\[NiftiMasker.wrapped] Resampling mask
________________________________________________________________________________
[Memory] Calling nilearn.image.resampling.resample_img...
resample_img(<nibabel.nifti1.Nifti1Image object at 0x7f8125fc1270>, target_affine=None, target_shape=None, copy=False, interpolation='nearest')
_____________________________________________________resample_img - 0.0s, 0.0min
\[NiftiMasker.wrapped] Finished fit
/home/runner/work/nilearn/nilearn/examples/03_connectivity/plot_seed_to_voxel_correlation.py:106: FutureWarning:
boolean values for 'standardize' will be deprecated in nilearn 0.15.0.
Use 'zscore_sample' instead of 'True' or use 'None' instead of 'False'.
________________________________________________________________________________
[Memory] Calling nilearn.maskers.nifti_masker.filter_and_mask...
filter_and_mask('/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_space-MNI152NLin2009cAsym_desc-preproc_bold.nii.gz',
<nibabel.nifti1.Nifti1Image object at 0x7f8125fc1270>, { 'clean_args': None,
'clean_kwargs': {},
'cmap': 'gray',
'detrend': True,
'dtype': None,
'high_pass': 0.01,
'high_variance_confounds': False,
'low_pass': 0.1,
'reports': True,
'runs': None,
'smoothing_fwhm': 6,
'standardize': False,
'standardize_confounds': True,
't_r': 2,
'target_affine': None,
'target_shape': None}, memory_level=1, memory=Memory(location=nilearn_cache/joblib), verbose=1, confounds=[ '/home/runner/work/nilearn/nilearn/nilearn_data/development_fmri/development_fmri/sub-pixar123_task-pixar_desc-reducedConfounds_regressors.tsv'], sample_mask=None, copy=True, sklearn_output_config=None)
\[NiftiMasker.wrapped] Loading data from <nibabel.nifti1.Nifti1Image object at
0x7f8125fc01c0>
\[NiftiMasker.wrapped] Smoothing images
\[NiftiMasker.wrapped] Extracting region signals
\[NiftiMasker.wrapped] Cleaning extracted signals
/home/runner/work/nilearn/nilearn/examples/03_connectivity/plot_seed_to_voxel_correlation.py:106: FutureWarning:
boolean values for 'standardize' will be deprecated in nilearn 0.15.0.
Use 'zscore_sample' instead of 'True' or use 'None' instead of 'False'.
_________________________________________________filter_and_mask - 11.2s, 0.2min
We can now inspect the extracted time series. Note that the seed time series is an array with shape n_volumes, 1), while the brain time series is an array with shape (n_volumes, n_voxels).
print(f"Seed time series shape: ({seed_time_series.shape})")
print(f"Brain time series shape: ({brain_time_series.shape})")
Seed time series shape: ((168, 1))
Brain time series shape: ((168, 32504))
We can plot the seed time series.
import matplotlib.pyplot as plt
plt.figure(constrained_layout=True)
plt.plot(seed_time_series)
plt.title("Seed time series (Posterior cingulate cortex)")
plt.xlabel("Scan number")
plt.ylabel("Normalized signal")
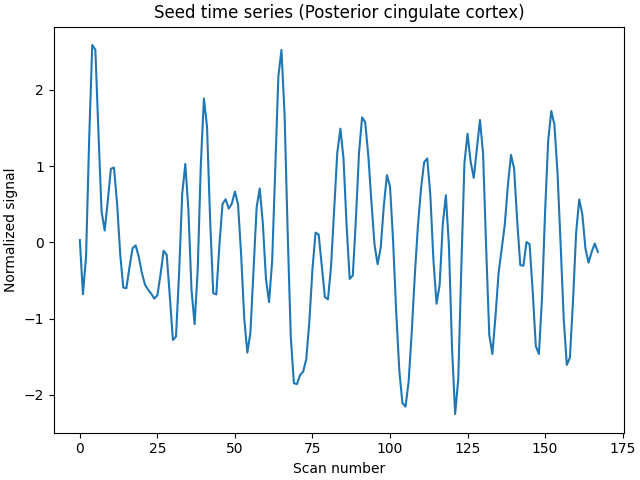
Text(18.166999999999994, 0.5, 'Normalized signal')
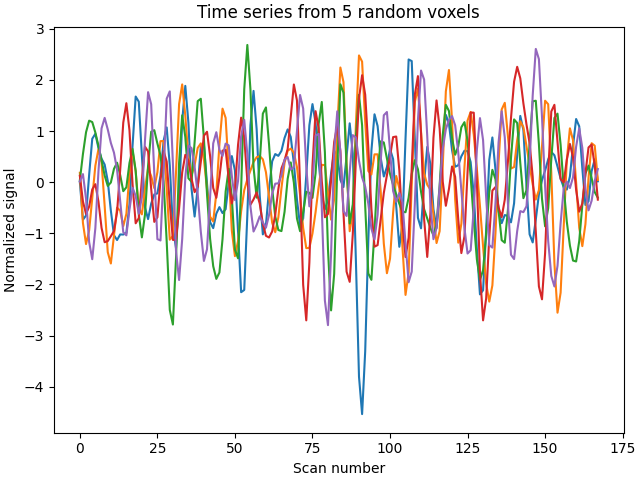
Exemplarily, we can also select 5 random voxels from the brain-wide data and plot the time series from.
plt.figure(constrained_layout=True)
plt.plot(brain_time_series[:, [10, 45, 100, 5000, 10000]])
plt.title("Time series from 5 random voxels")
plt.xlabel("Scan number")
plt.ylabel("Normalized signal")
plt.show()
Performing the seed-to-voxel correlation analysis¶
Now that we have two arrays (sphere signal: (n_volumes, 1), brain-wide voxel-wise signal (n_volumes, n_voxels)), we can correlate the seed signal with the signal of each voxel. The dot product of the two arrays will give us this correlation. Note that the signals have been variance-standardized during extraction. To have them standardized to norm unit, we further have to divide the result by the length of the time series.
import numpy as np
seed_to_voxel_correlations = (
np.dot(brain_time_series.T, seed_time_series) / seed_time_series.shape[0]
)
The resulting array will contain a value representing the correlation values between the signal in the seed region of interest and each voxel’s signal, and will be of shape (n_voxels, 1). The correlation values can potentially range between -1 and 1.
print(
"Seed-to-voxel correlation shape: ({}, {})".format(
*seed_to_voxel_correlations.shape
)
)
print(
f"Seed-to-voxel correlation: "
f"min = {seed_to_voxel_correlations.min():.3f}; "
f"max = {seed_to_voxel_correlations.max():.3f}"
)
Seed-to-voxel correlation shape: (32504, 1)
Seed-to-voxel correlation: min = -2.590; max = 5.146
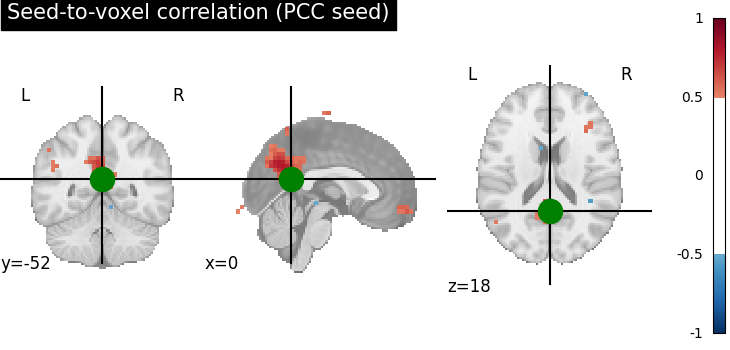
Plotting the seed-to-voxel correlation map¶
We can now plot the seed-to-voxel correlation map and perform thresholding to only show values more extreme than +/- 0.5. Before displaying, we need to create an in memory Nifti image object. Furthermore, we can display the location of the seed with a sphere and set the cross to the center of the seed region of interest.
seed_to_voxel_correlations_img = brain_masker.inverse_transform(
seed_to_voxel_correlations.T
)
display = plotting.plot_stat_map(
seed_to_voxel_correlations_img,
threshold=0.5,
vmax=1,
cut_coords=pcc_coords[0],
title="Seed-to-voxel correlation (PCC seed)",
)
display.add_markers(
marker_coords=pcc_coords, marker_color="g", marker_size=300
)
# At last, we save the plot as pdf.
from pathlib import Path
output_dir = Path.cwd() / "results" / "plot_seed_to_voxel_correlation"
output_dir.mkdir(exist_ok=True, parents=True)
print(f"Output will be saved to: {output_dir}")
display.savefig(output_dir / "pcc_seed_correlation.pdf")
\[NiftiMasker.inverse_transform] Computing image from signals
________________________________________________________________________________
[Memory] Calling nilearn.masking.unmask...
unmask(array([[0.014454, ..., 0.000316]]), <nibabel.nifti1.Nifti1Image object at 0x7f8125fc1270>)
___________________________________________________________unmask - 0.0s, 0.0min
Output will be saved to: /home/runner/work/nilearn/nilearn/examples/03_connectivity/results/plot_seed_to_voxel_correlation
Fisher-z transformation and save nifti¶
Finally, we can Fisher-z transform the data to achieve a normal distribution. The transformed array can now have values more extreme than +/- 1.
seed_to_voxel_correlations_fisher_z = np.arctanh(seed_to_voxel_correlations)
print(
"Seed-to-voxel correlation Fisher-z transformed: "
f"min = {seed_to_voxel_correlations_fisher_z.min():.3f}; "
f"max = {seed_to_voxel_correlations_fisher_z.max():.3f}f"
)
/home/runner/work/nilearn/nilearn/examples/03_connectivity/plot_seed_to_voxel_correlation.py:208: RuntimeWarning:
invalid value encountered in arctanh
Seed-to-voxel correlation Fisher-z transformed: min = nan; max = nanf
Eventually, we can transform the correlation array back to a Nifti image object, that we can save.
seed_to_voxel_correlations_fisher_z_img = brain_masker.inverse_transform(
seed_to_voxel_correlations_fisher_z.T
)
seed_to_voxel_correlations_fisher_z_img.to_filename(
output_dir / "pcc_seed_correlation_z.nii.gz"
)
\[NiftiMasker.inverse_transform] Computing image from signals
________________________________________________________________________________
[Memory] Calling nilearn.masking.unmask...
unmask(array([[0.014455, ..., 0.000316]]), <nibabel.nifti1.Nifti1Image object at 0x7f8125fc1270>)
___________________________________________________________unmask - 0.0s, 0.0min
Total running time of the script: (0 minutes 19.214 seconds)
Estimated memory usage: 640 MB